TL;DR
These object-oriented contstrained classes saved me so many times, that I can’t imagine my life without them. If it is good enough for the Bioconductor community to keep develop and maintain, there must be something very usefull in it, for other disciplines to learn.
Motivation
Multi-omics data integration and analysis. What a beast! It is one of the major challenges in the era of personalized/precision medicine (or whatever you want to call it). Denfinetely mine, as someone who is expected to grab such messy data from multiple sources, allign and annotate it altogether, and then testing some interesting hypothesis with it. If you thought analysis of a single omic (microarray, RNAseq etc) is overwhelming, what about integration of multiple single-omic data? Multi-omics data adds another layer of priceless high-throughput biologic data, attempting to reveal a snapshot from just another ‘biological’ angle, and another, and another… A typical single-omic data usually has two-dimensional matrix representation of measuring the intensity of thousand of genes/analytes (rows) for multiple subjects (columns). A multi-omic approach adds a third dimension, some sort of a data cube, of multiple layers of omics, each omic of different analytes (for the same subjects). That is a lot of data to organize and store. There must be a good, efficient systematic way to do it.

image credit: Nature
Designated R class to contain specific data
My first optometrist was not so much up to date with current technology (bear with me). In a standard eye exam, an optometrist tries many lenses, each has different characteristics, using an algorithm that should match the best combinations of lenses to fit the client’s very specific condition. This algorithm involves multiple iteration of trial and error, iteratively adjusting many lenses. I admire my optometrist. His job is definitely not easy. Not only he had to be familiar with all of the parameters, and the delicate complex algorithm, he also needed a good system to store the lenses in a way that is systematically accessible though this exhaustive search. I wondered how does he keep his lenses in order, and know exactly which one to pull, without having to go through all of them each time? Lucky for him, he had a good system for his lenses. They were stored in a well crafted customized cabinet that was designed for this very specific need.

Realization#1: Complex systems require designated containers! When designing these tools, one should consider the functionality of the common tasks that are operated with the stored components.
Back to our multi-omics data… Designing the tool (container) for such data should attempt to enable the following common tasks: Sorting, Annotating, Accessing (update: get/pull data, set/push data), Subsetting/slicing, to name just a few. Additional characterists to ease the performance should also have these design characteristics: Modularity, Automate authentication via self diagnosis / validation, Portability: self-contained, Extendiblity, Evolvability, Maintainability, Scalability, and above all, Simplicity (to some extent).

Designing a tool for handling such complex data and tasks can be done in R with simple tolls such as a ‘list of a list (of a list, of a list …)’. Well, good luck… been there, done that, learned my lessons.

There must be a better solution… I wish there would be a methodology that enables a system to be modeled as a set of objects that can be controlled and manipulated in a modular manner. This is what Object Oriented Programming (OOP) attempt to achieve, and R has some great tools to implement it.
Realization #2: No need to reinvent the wheel. Someone else, a well crafted expert in such task has already scalped such designated complex tool for all of us to utilize.
Why S4 ?
The Bioconductor community has been analyzing omics data for more than a decade, and over the time has scalped a well designated OOP R class to ease the handling and analysis of such data. The traditional R implemntation of OOD is the S3 class, but for reasons to be explained next, there was a need for some improvements, to accomplish such task. Hmmm…. How should we call the next generation of S3… surprise surprise… S4 ! . The architecture of this class has specific slots and methods (functions) that supports the routinely tasks of storing and handling of the data. In a first sight it may look overwhelming, and not much accessible, but it worth the time of learning to use its hidden gems.
The main idea of this class is to have a designated container for the raw data, that will also include a direct mapping to subject (column) annotation and gene/analyte (row) annotation. This comes very handy specially for the common task of subsetting the data. Slicing subsets of rows and/or columns does not have to be done multiple times at each of the relevant slots. Using the square brackets with the S4 class, one can easily slice through more than one slot, both vertically and horizontally, in a single operation: S4.class[ genes, subjects]
Unlike the S3 class, which access only the first attribute, S4 class implementation can access more than one slot. (Though S4 has other complexities that makes it not as much trivial/intuitive as the S3).
The evolvability of the S4 class for omic data
My mom once told me that all parents do some mistakes with their elder child, but the good news is that they do less mistakes with the next child. (Both of my parents, and myself are the elder child).
The S4 class for omics data also had its own learning curve. First came the eSet class, for the microarray assay data, but apparently it had some issues, so it is now deprecated. Then came the ExpressionSet class, with longer name and better support for the single omic data.
While new omic technologies emerged, some generalization of the former class had to be done, and it was evolved into a new S4 class, with even longer name, called SummarizedExperiment. This class also supports ranges of genomic sequences such as GenomicRanges (IRanges, Granges, Rle). The assay(s) slot of the SummarizedExperiment allow multiple matrices but of the same rows (analytes/genes).
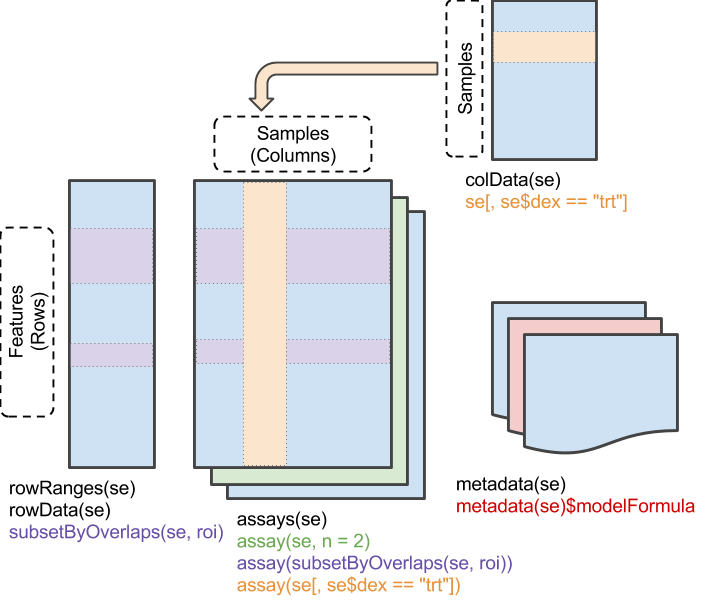
image credit: Bioconductor.org
But even that class could not catch up with the demanding need of the Bioconductor community. Different omic technologies has different analytes. We need a new class to handle experiments of multi-assays… How should we call the S4 class to handle that type of data…. MultiAssayExperiment.
The MultiAssayExperiment class has a third dimension, which allows a virtual stacking of each omic layer on top of each other, in parallel for each subject. This S4 object was extended with a third dimension: MultiAssayExperiment.class[ genes, subjects , Assays]
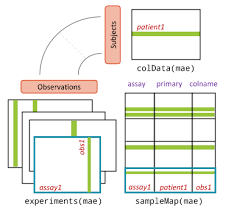
image credit: Bioconductor.org
The square brackets has a special feature which might be either annoying, or a bless, depends how well you utilize it. You probably think what if any of the subsetting return a single outcome (gene, subject or assay), or even an empty one? Aha… The drop=TRUE/FALSE magic parameter can take care of that to decide if the data.frame should be coerced into a vector, or maintain its data frame format. In addition, there is an internal mechanism of a set of self-validation rules that monitor and report a summary of which assays/analytes/subjects are stored.
The MultiAssayExperiment also has specialized methods (functions) for handling common tasks for such data. This include constructors, accessors (‘getters’, ‘setters’), management (handling replicate subjects), reshaping (wrangling the data into either long or wide format), and combining.
Conclusions:
Good design is inherited and carried over when the tool is redesigned to address new challenges. A solution may be good enough for a specific need, but when another layer is added, and dimension increase, a better design is required to address the new needs. It make sense for the new tool to inherit and maintain the good components from the previous tool, and relax/solve/modify/add the new elements that are needed. As new technologies emerge, and new layers are added, new tools are developed to address such challenges.
What’s next?
What is the next challenge with such data? I hope I didn’t lost you by now, so let’s review what we had so far:
A single-assay has many genes, for many subjects (ExpressionSet)
A (single) multi-assay experiment has multiple assays, for many subjects(MultiAssayExperiment).
but what if… I have multiple multi-assay data, e.g. TCGA, or CCLE projects that collect multi-omic data across many centers and for may different diseases types… This require a separate dedicated post.
Which other object oriented approach can achieve such functionality? How would R6 class address it?
Check more related topics here: https://drorberel.github.io/