
R package to bridge between Bioconductor’s S4 complex genomic data container, to mlr, a meta machine learning aggregator package.
Bioc2mlr is designed to convert Bioconductor S4 assay data containers summarizedExperiment, MultiAssayExperiment into generalized machnine learning environment.
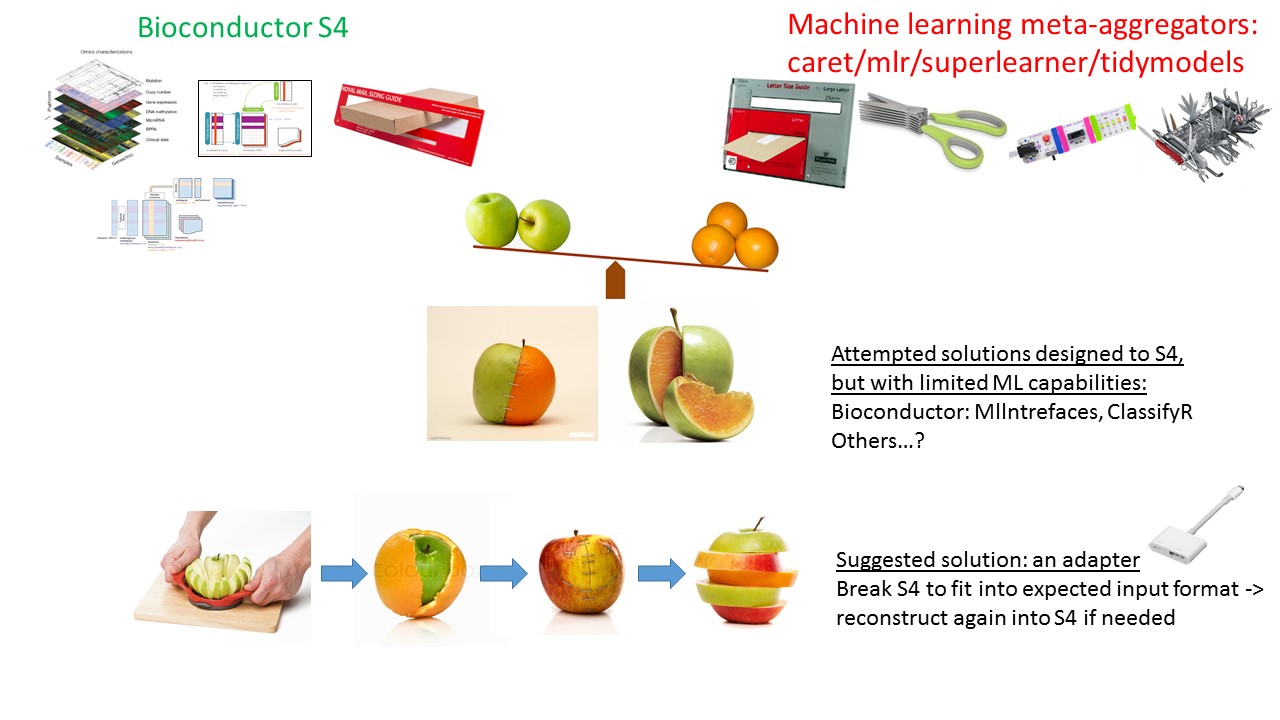
Bioconductor’s S4 data containers for genomic assays are popular, well established data structures. Their data architecture facilitates the application of common analytical procedures and well established statistical methodologies to large assay data. They are extensible to encompass new emerging technologies and analytical methods. However, the S4 system enforces strict constraints on the data and these constraints raise barriers for interoperability and integration with software and packages outside of Bioconductor’s repository.
mlr is a comprehensive package for machine learning. It aggregates hundreds of supervised and unsupervised models and facilitates analytics such as resampling, benchmarking, tuning, and ensemble. The mlrCPO package extends mlr’s pre-processing and feature engineering functionality via composable Preprocessing Operators (CPO) ‘pipelines’.
Bioc2mlr is a compact utility package designed to bridge between these approaches. It deploys transformations of SummarizedExperiment and MultiAssayExperiment S4 data structures into mlr’s expected format. It also implements Bioconductor’s popular feature selection (filtering) methods used by limma package and others, as a CPO. The vignettes present comparisons to the MLInterfaces package, which aims to achieve similar goals, and presents workflows for popular publicly available genomic datasets such as curatedTCGAData.
Current implementations
Two Bioconductor assay container are currently implemented: SummarizedExperiment for a single assay (though may have multiple sub-assays slots), and MultiAssayExperiment for multiple assays. Within the machine-learning framework, the two main steps that are adapted are the pre-processing step, followed by the (multivariate) model fitting.
Tools will be demonstrated for each of these 4 combinations.
| S4 assay data container | Pre-processing (TBA) | Model (multivariate) |
|---|---|---|
| SummarizedExperiment (SE) | limmaCPO | Fun_SE_to_taskFunc |
| MultiAssayExperiment (MAE) | UnivCPO | Fun_MAE_to_taskFunc |
Model-evaluation (ML)
A. SummarizedExperiment (SE)
Convert raw data from SE S4 class, to mlr’s “task”
data(Golub_Merge, package = 'golubEsets') # ExpressionSet
smallG<-Golub_Merge[200:259,]
smallG
#> ExpressionSet (storageMode: lockedEnvironment)
#> assayData: 60 features, 72 samples
#> element names: exprs
#> protocolData: none
#> phenoData
#> sampleNames: 39 40 ... 33 (72 total)
#> varLabels: Samples ALL.AML ... Source (11 total)
#> varMetadata: labelDescription
#> featureData: none
#> experimentData: use 'experimentData(object)'
#> pubMedIds: 10521349
#> Annotation: hu6800
library(SummarizedExperiment)
smallG_SE<-makeSummarizedExperimentFromExpressionSet(smallG)
# functional:
task_SE_Functional<-Fun_SE_to_taskFunc(smallG_SE, param.Y.name = 'ALL.AML', param.covariates = NULL, param_positive_y_level = 'ALL', task_return_format = 'functional', task_type = 'classif') ## will work with either 1 or multiple assayS
task_SE_Functional
#> Supervised task: DF_functionals
#> Type: classif
#> Target: ALL.AML
#> Observations: 72
#> Features:
#> numerics factors ordered functionals
#> 0 0 0 1
#> Missings: FALSE
#> Has weights: FALSE
#> Has blocking: FALSE
#> Has coordinates: FALSE
#> Classes: 2
#> ALL AML
#> 47 25
#> Positive class: ALL
# non-functional:
## 1. directly, but into DF
extracted_DF_from_task_SE<-getTaskData(task_SE_Functional, functionals.as = "dfcols") # keep matrix
extracted_DF_from_task_SE[,1:10] %>% str
#> 'data.frame': 72 obs. of 10 variables:
#> $ ALL.AML : Factor w/ 2 levels "ALL","AML": 1 1 1 1 1 1 1 1 1 1 ...
#> $ exprs.D13627_at: num 330 544 978 1035 3895 ...
#> $ exprs.D13628_at: num 229 147 110 237 106 256 144 84 -7 -3 ...
#> $ exprs.D13630_at: num 359 289 609 485 866 663 673 401 480 273 ...
#> $ exprs.D13633_at: num -9 57 207 302 475 0 112 257 244 252 ...
#> $ exprs.D13634_at: num 115 248 91 58 244 245 98 182 186 241 ...
#> $ exprs.D13635_at: num 31 -43 40 31 84 -159 -7 -2 62 111 ...
#> $ exprs.D13636_at: num 195 23 -60 317 449 -262 386 295 177 51 ...
#> $ exprs.D13637_at: num 161 137 -94 -96 432 -535 136 86 99 143 ...
#> $ exprs.D13639_at: num 456 3336 655 2771 3575 ...
## 2. Fun_SE_to_taskFunc(..., task_return_format = 'dfcols')
task_SE_NON_Functional<-Fun_SE_to_taskFunc(smallG_SE, param.Y.name = 'ALL.AML', param.covariates = NULL, param_positive_y_level = 'ALL', task_return_format = 'dfcols', task_type = 'classif') ## will work with either 1 or multiple assayS
## 3. functional_to_NonFunctional_task_function(task_functional)
task_SE_NON_Functional_alt<-functional_to_NonFunctional_task_function(task_SE_Functional)
## 4. designated function ## TBA
# extracted = extractFDAFeatures(task_SE_Functional, feat.methods = list("exprs" = all))Single assay ML demonstration
MLInterface
library(MLInterfaces)
#> Warning: package 'MLInterfaces' was built under R version 3.5.1
#> Warning: package 'XML' was built under R version 3.5.2
krun<-MLearn(formula = ALL.AML~., data = smallG, .method = knnI(k=1), trainInd = 1:40)
krun
#> MLInterfaces classification output container
#> The call was:
#> MLearn(formula = ALL.AML ~ ., data = smallG, .method = knnI(k = 1),
#> trainInd = 1:40)
#> Predicted outcome distribution for test set:
#>
#> ALL AML
#> 22 10
#> Summary of scores on test set (use testScores() method for details):
#> Min. 1st Qu. Median Mean 3rd Qu. Max.
#> 1 1 1 1 1 1
confuMat(krun)
#> predicted
#> given ALL AML
#> ALL 18 3
#> AML 4 7mlr
task_train<-task_SE_Functional %>% subsetTask(subset = 1:40)
task_test <-task_SE_Functional %>% subsetTask(subset = 41:72)
classif.lrn = makeLearner("classif.knn")
model<-train(classif.lrn, task_train)
Predict<-model %>% predict(task_test)
Predict %>% calculateConfusionMatrix()
#> predicted
#> true ALL AML -err.-
#> ALL 18 3 3
#> AML 4 7 4
#> -err.- 4 3 7B. MultiAssayExperiment (MAE)
Two data examples:
1. miniACC, balanced, without ‘dropouts’.
2. Customized, non-balanced, with ‘dropouts’.
Convert raw data from MAE S4 class, to mlr’s “task”
1. miniACC
library(MultiAssayExperiment)
miniACC
#> A MultiAssayExperiment object of 5 listed
#> experiments with user-defined names and respective classes.
#> Containing an ExperimentList class object of length 5:
#> [1] RNASeq2GeneNorm: SummarizedExperiment with 198 rows and 79 columns
#> [2] gistict: SummarizedExperiment with 198 rows and 90 columns
#> [3] RPPAArray: SummarizedExperiment with 33 rows and 46 columns
#> [4] Mutations: matrix with 97 rows and 90 columns
#> [5] miRNASeqGene: SummarizedExperiment with 471 rows and 80 columns
#> Features:
#> experiments() - obtain the ExperimentList instance
#> colData() - the primary/phenotype DataFrame
#> sampleMap() - the sample availability DataFrame
#> `$`, `[`, `[[` - extract colData columns, subset, or experiment
#> *Format() - convert into a long or wide DataFrame
#> assays() - convert ExperimentList to a SimpleList of matrices
# miniACC %>% sampleMap %>% data.frame %>% dplyr::select(primary, assay) %>% table # no replicates within same assay
task_Functional_MAE<-Fun_MAE_to_taskFunc(miniACC, param.Y.name = 'vital_status', param.covariates = c('gender','days_to_death'), param_positive_y_level = '1', task_type = 'classif')
task_Functional_MAE
#> Supervised task: DF_functionals
#> Type: classif
#> Target: vital_status
#> Observations: 385
#> Features:
#> numerics factors ordered functionals
#> 1 5 0 5
#> Missings: TRUE
#> Has weights: FALSE
#> Has blocking: FALSE
#> Has coordinates: FALSE
#> Classes: 2
#> 0 1
#> 248 137
#> Positive class: 1
extracted_DF_from_task_MAE_functionals<-getTaskData(task_Functional_MAE, functionals.as = "matrix") # keep functionals
extracted_DF_from_task_MAE_functionals[,1:10] %>% glimpse
#> Observations: 385
#> Variables: 10
#> $ Unique_sample_id <fct> RNASeq2GeneNorm_TCGA-OR-A5J1_TCGA-OR-A5J1-01A...
#> $ assay <fct> RNASeq2GeneNorm, RNASeq2GeneNorm, RNASeq2Gene...
#> $ primary <fct> TCGA-OR-A5J1, TCGA-OR-A5J2, TCGA-OR-A5J3, TCG...
#> $ colname <fct> TCGA-OR-A5J1-01A-11R-A29S-07, TCGA-OR-A5J2-01...
#> $ gender <fct> male, female, female, male, female, female, m...
#> $ days_to_death <int> 1355, 1677, NA, 365, NA, 490, 579, NA, 922, 5...
#> $ vital_status <fct> 1, 1, 0, 1, 0, 1, 1, 0, 1, 1, 1, 0, 1, 0, 1, ...
#> $ RNASeq2GeneNorm <dbl> <matrix[25 x 198]>
#> $ gistict <dbl> <matrix[25 x 198]>
#> $ RPPAArray <dbl> <matrix[25 x 33]>
extracted_DF_from_task_MAE_dfcols<-getTaskData(task_Functional_MAE, functionals.as = "dfcols") # concatonate functionals
extracted_DF_from_task_MAE_dfcols[,1:10] %>% glimpse
#> Observations: 385
#> Variables: 10
#> $ Unique_sample_id <fct> RNASeq2GeneNorm_TCGA-OR-A5J1_TCGA-OR-A5...
#> $ assay <fct> RNASeq2GeneNorm, RNASeq2GeneNorm, RNASe...
#> $ primary <fct> TCGA-OR-A5J1, TCGA-OR-A5J2, TCGA-OR-A5J...
#> $ colname <fct> TCGA-OR-A5J1-01A-11R-A29S-07, TCGA-OR-A...
#> $ gender <fct> male, female, female, male, female, fem...
#> $ days_to_death <int> 1355, 1677, NA, 365, NA, 490, 579, NA, ...
#> $ vital_status <fct> 1, 1, 0, 1, 0, 1, 1, 0, 1, 1, 1, 0, 1, ...
#> $ RNASeq2GeneNorm.DIRAS3 <dbl> 1487.0317, 9.6631, 18.9602, 760.6507, 1...
#> $ RNASeq2GeneNorm.MAPK14 <dbl> 778.5783, 2823.6469, 1061.7686, 806.351...
#> $ RNASeq2GeneNorm.YAP1 <dbl> 1009.6061, 2305.0590, 1561.2502, 713.40...2. Customized
library(MultiAssayExperiment)
patient.data <- data.frame(sex=c("M", "F", "M", "F", "F"),
age=38:42,
row.names=c("Jack", "Jill", "Bob", "Barbara","Meg"))
## assay A
arraydat <- matrix(seq(101, 108), ncol=4,
dimnames=list(c("ENST00000294241", "ENST00000355076"),
c("array1", "array2", "array3", "array4")))
coldat <- data.frame(slope53=rnorm(4), row.names=c("array1", "array2", "array3", "array4"))
exprdat <- SummarizedExperiment(arraydat, colData=coldat)
exprmap <- data.frame(primary=c("Jill", "Jill", "Meg", "Barbara"),
colname=c("array1", "array2", "array3", "array4"),
stringsAsFactors = FALSE)
## assay B
methyldat <-
matrix(1:10, ncol=5,
dimnames=list(c("ENST00000355076", "ENST00000383706"),
c("methyl1", "methyl2", "methyl3",
"methyl4", "methyl5")))
methylmap <- data.frame(primary = c("Jack", "Jack", "Jack", "Meg", "Bob"),
colname = c("methyl1", "methyl2", "methyl3", "methyl4", "methyl5"),
stringsAsFactors = FALSE)
myMultiAssay <- MultiAssayExperiment(list("A" = exprdat, "B" = methyldat), patient.data, list(A = exprmap, B = methylmap) %>% listToMap)
myMultiAssay
#> A MultiAssayExperiment object of 2 listed
#> experiments with user-defined names and respective classes.
#> Containing an ExperimentList class object of length 2:
#> [1] A: SummarizedExperiment with 2 rows and 4 columns
#> [2] B: matrix with 2 rows and 5 columns
#> Features:
#> experiments() - obtain the ExperimentList instance
#> colData() - the primary/phenotype DataFrame
#> sampleMap() - the sample availability DataFrame
#> `$`, `[`, `[[` - extract colData columns, subset, or experiment
#> *Format() - convert into a long or wide DataFrame
#> assays() - convert ExperimentList to a SimpleList of matrices
myMultiAssay %>% sampleMap %>% data.frame %>% select(primary, assay) %>% table # Yes replicates within same assay, and non-balanced / dropouts!!!
#> assay
#> primary A B
#> Barbara 1 0
#> Bob 0 1
#> Jack 0 3
#> Jill 2 0
#> Meg 1 1
# myMultiAssay %>% sampleMap %>% data.frame %>% filter(assay == 'A')
# myMultiAssay$sex
task_Functional_MAE_customized<-Fun_MAE_to_taskFunc(myMultiAssay, param.Y.name = 'sex', param.covariates = NULL, param_positive_y_level = 'M', task_type = 'classif')Multi-assay ML demonstration
mlr: vertical integration
Unless the learner has sepecific implementation for functional data, it will be automatically converted into standard (non-functional) task.
bartMachine model was chosed only because it has a built-in NA handling. Any other ‘learner’ from mlr could be demonstrated instead.
library(bartMachine)
classif_lrn_bartMachine<-makeLearner("classif.bartMachine")
model_bartMachine<-train(classif_lrn_bartMachine, task_Functional_MAE)
#> bartMachine initializing with 50 trees...
#> bartMachine vars checked...
#> bartMachine java init...
#> bartMachine factors created...
#> bartMachine before preprocess...
#> bartMachine after preprocess... 1868 total features...
#> warning: cannot use MSE of linear model for s_sq_y if p > n. bartMachine will use sample var(y) instead.
#> bartMachine sigsq estimated...
#> bartMachine training data finalized...
#> Now building bartMachine for classification ...Covariate importance prior ON. Missing data feature ON.
#> evaluating in sample data...done
Predict_bartMachine<-model_bartMachine %>% predict(task_Functional_MAE)
Predict_bartMachine %>% calculateConfusionMatrix()
#> predicted
#> true 0 1 -err.-
#> 0 248 0 0
#> 1 0 137 0
#> -err.- 0 0 0Case studies (TBA):
1. CAVDmetaMAE: proof-of-concept example
CAVD dataspace is an online resouce to access and analyze HIV vaccine experimental assay data. It is annotated, and accessible via either online tool, and R API DataSpaceR.
The CAVDmetaMAE package implement a hypothesis-free approach, to find best candidates of immune biomarkers, that are associated with experimental groups, at each study (separately), and across all studies together (meta-analysis).
Within each study, immune biomarkers will be analyzed by both single assays, and combinations across multiple assays.
https://github.com/drorberel/CAVDmetaMAE
Private repo. Access permission by request.
2. Multi-assay customized feature selection for JDRF data (under review)
3. Annotated public datasets (TBA)
TCGA curatedTCGAData
Microbiome curatedMetagenomicData